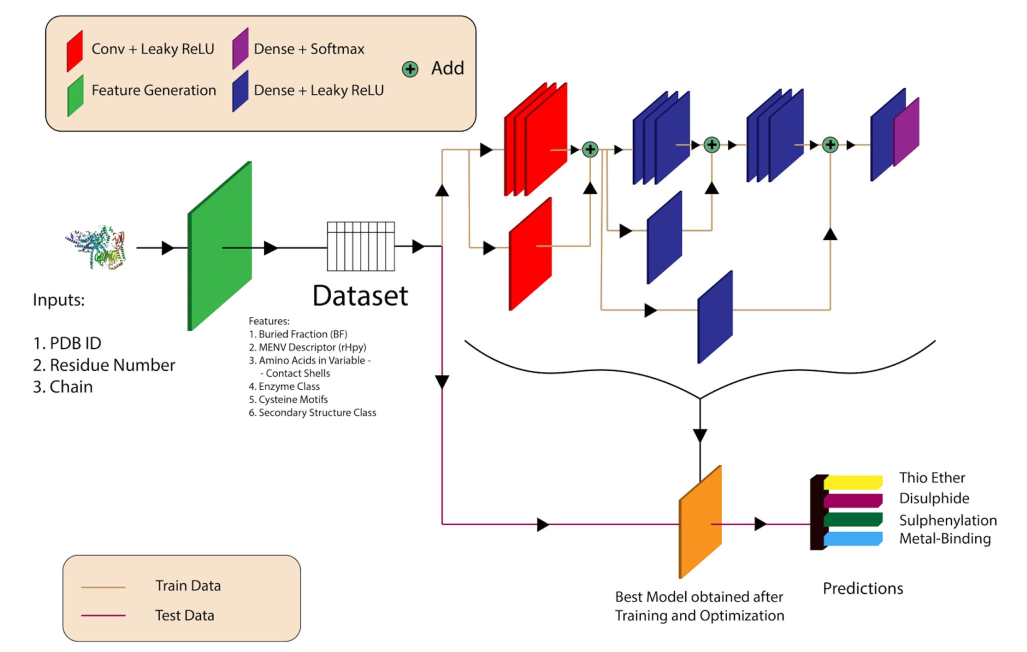

DeepCys: Structure-based multiple cysteine function prediction method trained on deep neural network: Case study on domains of unknown functions belonging to COX2 domains
Cysteine (Cys) is the most reactive amino acid participating in a wide range of biological functions. In-silico predictions complement the experiments to meet the need of functional characterization. Multiple Cys function prediction algorithm is scarce, in contrast to specific function prediction algorithms. Here we present a deep neural network-based multiple cysteine function prediction, available on web-server (DeepCys). DeepCys model was trained and tested on two independent datasets curated from protein crystal structures. This prediction method requires three inputs, namely, PDB identifier (ID), chain ID and residue ID for a given cysteine and outputs the probabilities of four cysteine modifications, namely, Disulphide, Metal-binding, Thioether and S-sulphenylation and predicts the most probable cysteine modification. The algorithm exploits the local and global protein properties, like, sequence and secondary structure motifs, buried fractions, microenvironments and protein/enzyme class. DeepCys outperformed most of the multiple and specific cysteine modification algorithms. This method can predict maximum number of cysteine modifications. Moreover, for the first time, explicitly predicts thioether function. This tool was used to elucidate the cysteine modifications on domains of unknown functions belonging to Cytochrome c oxidase subunit-II like transmembrane domains. Apart from the web-server, a standalone program is also available on GitHub.
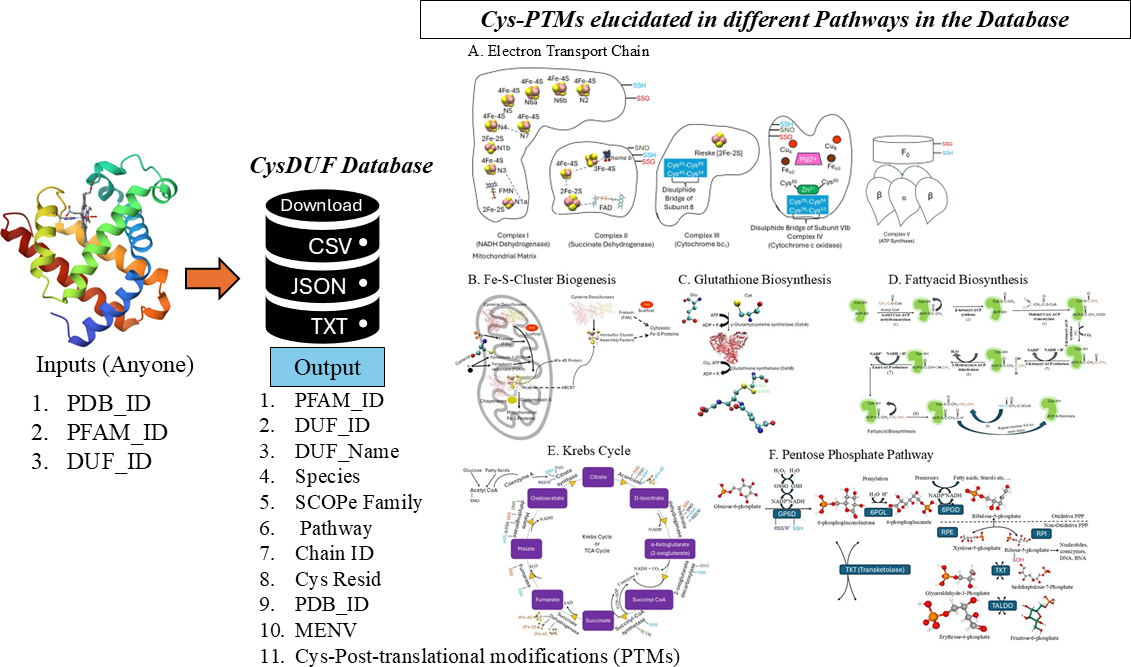
CysDuF database: annotation and characterization of Cysteine residues in Domain of Unknown Function (DUF) proteins based on Cysteine post-translational modifications, their protein microenvironments, biochemical pathways, taxonomy, and diseases
Experimental characterization and annotation of amino acids belonging to Domains of Unknown Function (DUF) proteins are expensive, and time-consuming which could be complemented by computational methods. Cysteine, being the second most reactive amino acid at the catalytic sites of enzymes, was selected for functional annotation and characterization on DUF proteins. Earlier we reported functional annotation of Cysteine on DUF proteins belonging to the COX-II family. However, holistic characterization of Cysteine functions on DUF proteins was not known, to the best of our knowledge. Here, we annotated and characterized Cysteine residues based on post-translational modifications (PTMs), biochemical pathways, diseases, taxonomy, and protein microenvironment. The information on uncharacterized DUF proteins was initially obtained from the literature and the sequence, structure, pathways, taxonomy, and disease information were retrieved from the SCOPe database using DUF IDs. Protein microenvironments (MENV) around Cysteine residues from DUF proteins were computed using protein structures (n=70342). The Cysteine PTMs were predicted using the in-house Cysteine-function prediction server, DeepCys . The accuracy of the prediction, validated against known experimental Cysteine PTMs (n=18626) was 0.79. The information was consolidated in the database, retrievable in downloadable formats (CSV, JSON, or TXT) using the following inputs, DUF ID, PFAM ID, or PDB ID. For the first time, we annotated Cysteine PTMs in DUF proteins belonging to seven different biochemical pathways and various species across the taxonomy, notably for the SARS-COV2 virus. The nature of MENV around Cysteine from DUF proteins was mainly buried and hydrophobic. However, in the SARS-COV2 virus, a significant number of functional Cysteine residues were exposed on the surface with hydrophilic microenvironment.
SeqDeepCys: Sequence-based multiple cysteine function prediction method trained on artifical neural networks: Case study on SARS-CoV-2 and Mitocarta3.
Cysteine (Cys) amino acid undergoes multiple post-translational modifications due to its reactive thiol group. A wide range of biological functions make cysteine residue a crucial target for the prediction of different post-translational modifications in a given protein. There are a large number of web servers that predict only a handful of cysteine post-translational modifications and a limited number of servers predicting multiple modifications, including the one we published based on structure. Here we developed a prediction tool for multiple cysteine post-translational modifications based on the sequence. Prediction of thioether using sequence is reported here for the first time to the best of our knowledge. The proposed prediction tool will be able to predict seven post-translational modifications, namely, Thioether, Sulphenylation, S-Nitrosylation, S-Palmitoylation, S-Glutathionylation, Disulphide, and Metal-Binding . We present a machine learning and deep learning approach-based multiple Cys function prediction (SeqDeepCys) tool. The model was trained and tested on independent datasets curated from the protein sequences. The prediction accepts two inputs, namely FASTA sequences and Residue Number for a given Cys. The prediction outputs are the probabilities of seven cysteine post-translational modifications. The proposed method can predict a maximum number of cysteine functions. Moreover, for the first time, it explicitly predicts the thioether function. The developed prediction model is compared with the existing prediction web servers based on accuracy. In future, the prediction model will be deployed as a web server, and a stand-alone package.